A key challenge for the commercial viability of ATMPs is achieving both a high price and minimising the likelihood of reimbursement restrictions. The Parallel Consultation procedure provides consolidated feedback from European regulators (EMA) and a group of national health technology assessment (HTA) bodies on the data needed to get both regulatory approval and reimbursement.
The challenge
Established in July 2017, the Parallel Consultation procedure (facilitated by EMA in collaboration with EUnetHTA) provides an opportunity for medicine developers to obtain consolidated feedback from European regulators and a group of national health technology assessment (HTA) bodies to optimise their clinical development programme. This is to ensure it presents the best possible case for both regulatory approval and reimbursement. The fact that only a very limited number of developers have completed it so far means that very few stakeholders have first-hand experience of this procedure.
A challenge for a meaningful engagement with the HTA bodies specifically is that, unlike regulatory evidence requirements by EMA which have a binary outcome (securing or not securing market authorisation), HTA evidence requirements vary significantly for different target price and reimbursement scenarios.
Whereas an HTA recommendation for adoption may be secured on a given data set, what matters the most for a manufacturer is that it corresponds to a commercially viable price and volume i.e. size of qualifying patient population (HTA bodies can restrict reimbursement to a subpopulation of the target population described in the regulatory label). Therefore, in the context of the parallel consultation procedure, it is important to understand the evidence required for the reimbursement scenario that secures commercial viability rather than the evidence required for securing reimbursement in general. If these important dimensions are not understood in detail and considered strategically in both the development of briefing document and the engagement with HTA bodies, the value of pursuing the parallel consultation is limited.
HTA assessments follow value-based frameworks that rely on evidence of the value added over existing therapeutic alternatives. The therapy’s reimbursed price potential is determined by quantifying and monetising the magnitude of the added clinical benefit; however, the monetisation mechanism differs across markets.
ATMPs have high manufacturing costs that dictate a high target price in order to be commercially viable. To secure reimbursement at a commercially viable price, it is important to ensure that the therapy’s incremental cost to the healthcare system (above current therapeutic approaches) is justified by the incremental benefits it delivers.
ATMPs face two common challenges in generating evidence that supports the added benefit for the purpose of HTA:
- Robustly demonstrating incremental benefit over the standard of care may be challenging due to clinical feasibility constraints:
- Gold-standard head-to-head trial design may not be possible
- Randomised placebo-controlled trials may not be feasible (limiting the prospect for credible indirect comparisons)
- Robust comparative data from single arm trials may not be feasible due to a lack of historical control data, homogeneous patient populations, and/or where therapies are targeting indications where the natural history of disease is not well known
- Statistical significance can be constrained by small sample sizes, especially for therapies targeting rare diseases (where recruitment is challenging)
- The use of surrogate outcomes (rather than hard clinical outcomes) may impair the acceptability of magnitude of treatment effect; a recent report (NICE Regenerative Medicine Study, 2016) claims that the magnitude of effect may be overestimated when using surrogate outcomes
- In some cases of very rare diseases comparable treatment and measures of outcome may not be available
- Long-term claims may be difficult to substantiate through clinical data
- Lack of long-term data at launch can increase the uncertainty around long-term claims regarding maintenance of effect and safety
Given the challenges ATMPs are facing regarding supporting data, consultations with HTA bodies early in development are highly recommended so that evidence generation plans can be informed accordingly. Key topics for seeking advice from HTA bodies include:
- The most relevant clinical, patient-reported and economic outcomes
- The most relevant comparators
- Quantification of the minimal important difference in incremental benefit versus standard of care/best supportive care
- The most relevant economic models for a given jurisdiction
- Where H2H trials not feasible, agree alternatives to generating comparative data
- Where indirect comparisons will be leveraged, explore acceptability and methodological robustness
- Where only single arm trials are feasible, agree how historical controls may be leveraged
- Where surrogate endpoints will be used, agree selection and validation
- Where long term claims will be made beyond the duration of the clinical development programme, explore:
- Alternative data-sets
- Whether modelled data can be used to bridge the evidence gap
- Acceptable approaches for dealing with data uncertainty at the time of launch
- Where no comparable treatment and measures of outcome are available early engagement with EMA and HTA bodies will help manufacturers agree the development of appropriate outcome measures and approaches to substantiate incremental benefit claims
While the insights derived from the Parallel Consultation procedure can be of great strategic importance, the output depends greatly on the information the medicine developer provides in the briefing document for the consultation. In order to maximise benefits from the consultation with HTA bodies, it is important to conduct the following activities in a sequential manner:
- Understand value drivers for a given therapy and how these can help support a commercially viable price and volume opportunity (given a high manufacturing cost base); this information forms the basis for the development of the target value story
- Develop the briefing document for the parallel consultation; articulate clearly:
- The target value story
- The evidence generation plan that supports the target value story
- The areas where evidence gaps may exist; formulate questions for HTA bodies and propose potential solutions


The value story is a key component of the briefing document and details how the therapy is expected to address the unmet needs in the therapy area it is targeting. The top level value story typically includes an overarching paragraph that summarises the value proposition, followed by supporting value statements that relate to the unmet need in the target patient population and the clinical and economic implications for the patient and the healthcare system arising from adopting the novel therapy. The value story and supporting evidence directly impact the price and volume potential for a novel therapy; the numerical interrelationship between therapy value drivers, reimbursed price and volume opportunity can be determined through health-economic analysis and pricing research.
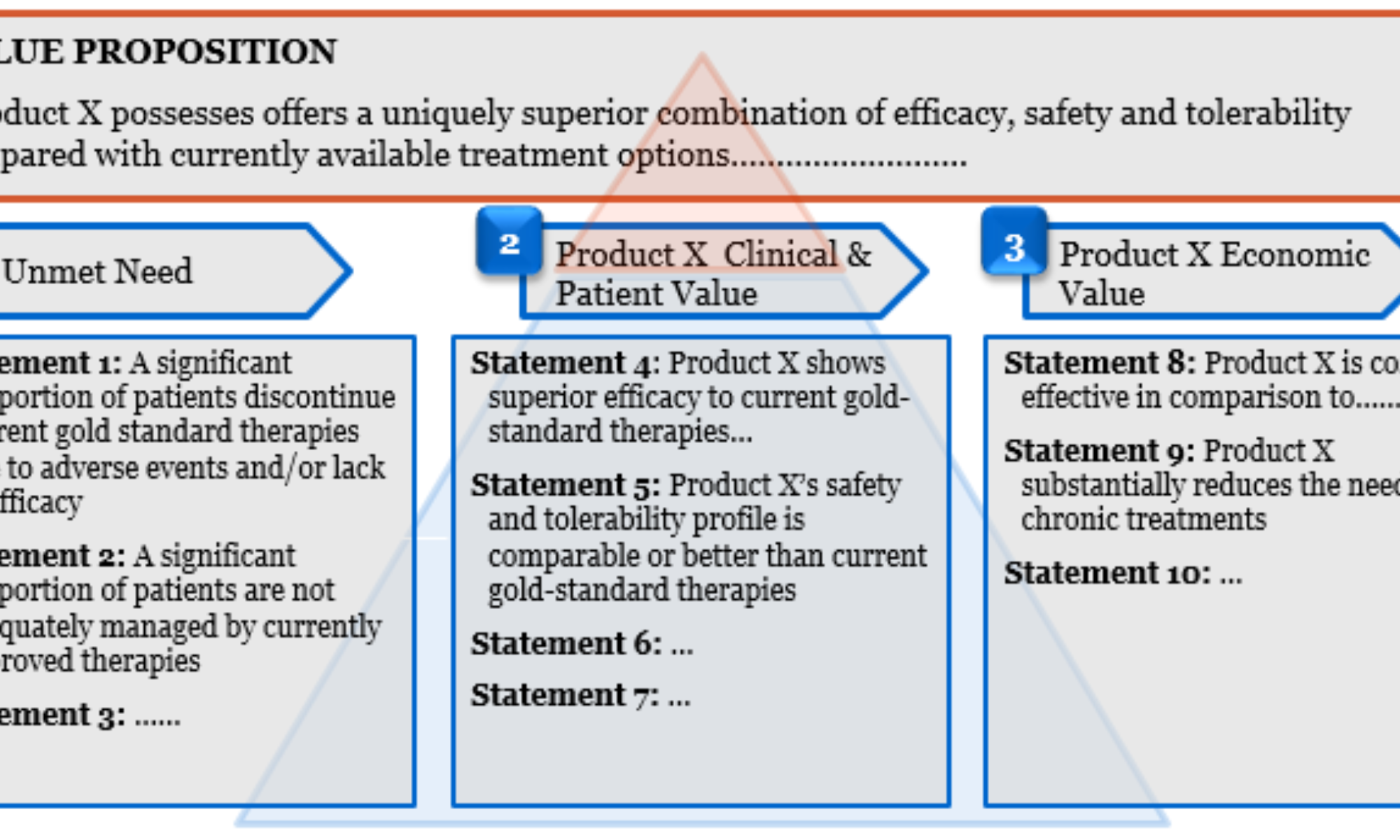
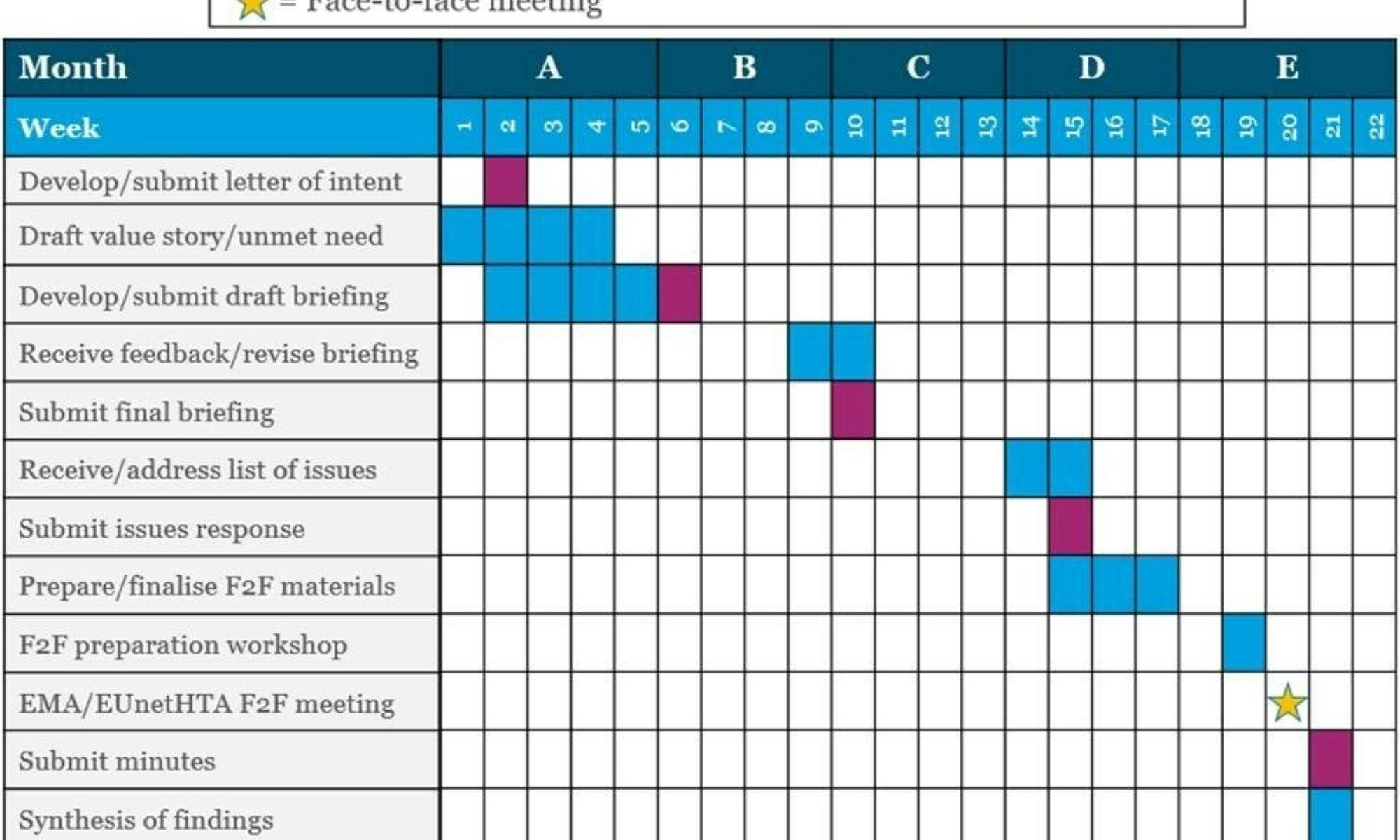
The process
The Parallel Consultation procedure takes around four months to complete, and is structured according to the EMA/EUnetHTA’s submission deadlines and key milestones:
- Submission of Letter of intent
- Submission of draft briefing document
- Submission of final briefing document
- Submission of written response to the EMA/EUnetHTA list of issues
- Participation at the face-to-face meeting with EMA/EUnetHTA
- Submission of minutes from the face-to-face meeting with EMA/EUnetHTA
The HE&MA team of the Cell and Gene Therapy Catapult is one of very few stakeholders to have completed the Parallel Consultation procedure, since its introduction in July 2017. Our experience and expertise uniquely positions us to ensure that the output developers obtain from the HTA bodies provide the insights needed to tailor the evidence generation plan to minimise commercial risks, and maximise their therapy’s adoption potential through:
- Helping ATMP developers identify the target price and volume that support commercial viability (where ATMP developers have not already done so)
- Guiding ATMP developers through the development of a value story that supports the target price in the target population
- Helping to shape the evidence generation plan to support the value story within the remits of clinical feasibility and regulatory requirements
- Identifying areas where proposed evidence generation plan may present HTA challenges (e.g. evidence gaps)
- Working with ATMP developers to identify potential solutions to these challenges, and use this information to formulate position statements in the briefing document to form the basis for a constructive dialogue with HTA bodies
- Helping developers address the list of issues that EMA/HTA bodies highlight following their review of the briefing document
- Helping to prepare and facilitate a constructive dialogue during the face-to-face consultation with EMA/HTA bodies
- Capturing learnings and developing strategic recommendations to shape the evidence generation plan
- Formulating an early pricing and reimbursement strategy leveraging our extensive experience in P&R for high cost innovative therapies and more specifically ATMPs
The team
The Health Economics and Market Access (HE&MA) function of the Cell and Gene Therapy Catapult is led by Panos Kefalas, who brings over 15 years’ experience in pharmaceutical pricing, reimbursement and health economics gained from senior roles with major HEOR and market access consultancy firms (IMS Health plc., PriceSpective / ICON plc., Evidera plc.) and from managing NICE guidance development. He is supported by an in-house team of experienced in health economics and market access professionals, each with a more than seven years’ experience in the HE&MA area.